Our research activities focus on the development of methods and workflows that utilize accurate quantum chemical methods together with artificial intelligence and machine learning. Our overall objectives are to elucidate the fundamental physical principles underlying chemical reactivity and catalysis, separation processes, and heavy element chemistry, as well as to assist in the interpretation of experimental data. Currently, our group is active on the following projects:
1. Data-driven Quantum Chemistry
Quantum chemistry is a branch of chemistry that applies quantum mechanics for the theoretical description of chemical phenomena. Wave function-based methods are nowadays used routinely for the calculation of electronic energies and properties of small molecules. However, the underlying complexity of the equations that have to be solved and the computational effort needed for qualitative results hinders the applicability to larger chemical systems or to the computation of potential energy surfaces. An approach that can surpass these bottlenecks is the combination of quantum chemistry with state-of-the-art machine learning algorithms. In a recent publication we presented a data-driven computational approach that can be applied for a large variety of electronic structure theory methods. In our methodology, we learn the electronic wave function from data obtained from small molecules. For demonstrating its applicability, we are examining the convergence of the coupled-cluster singles-and-doubles (CCSD) iterative solver. Our results show a remarkable speed-up for the convergence of the CCSD equations. The data-driven CCSD (DDCCSD) is not an alchemical method since the actual iterative coupled-cluster equations are solved. We anticipate extending this approach to electronic structure theory methods that capture the strong correlation of molecules.

Related Publications:
(1) P. D. V. S. Pathirage, J. T. Phillips, K. D. Vogiatzis Exploration of the Two-electron Excitation Space with Data-driven Coupled-cluster, J. Phys. Chem. A, 2024, 128, 1938.
(2) G. M. Jones, R. R. Li, A. E. DePrince III, K. D. Vogiatzis Data-driven Refinement of Electronic Energies from Two-electron Reduced-density-matrix Theory, J. Phys. Chem. Lett., 2023, 14, 6377.
(3) G. M. Jones, P. D. V. S. Pathirage, K. D. Vogiatzis, Data-driven Acceleration of Coupled-Cluster Theory and Perturbation Theory Methods, in: “Quantum Chemistry in the Age of Machine Learning”, 2022, Editor: Pavlo Dral, Elsevier, pp. 509-529.
(4) J. Townsend, K. D. Vogiatzis, Transferable MP2-Based Machine Learning for Accurate Coupled-Cluster Energies, J. Chem. Theory Comput., 2020, 16, 7453.
(5) J. Townsend, K. D. Vogiatzis, Data-Driven Acceleration of the Coupled-Cluster Singles and Doubles Iterative Solver, J. Phys. Chem. Lett., 2019, 10, 4129.
2. Gas Separations with Polymeric Membranes
Membrane-based gas separations are one of the most promising approaches for CO2 capture from the flue gas. Our aim is to design in silico the next generation of high-performance membranes with increased permeability and selectivity. We are interested on the exploration of the electronic effects that are important for gas separations with industrial relevance. In particular, non-covalent interactions between gas molecules and materials play an important role on such processes. We have developed novel tools that allow us to perform fast and reliable screening of large molecular databases for the discovery of molecules with stronger CO2 affinity.
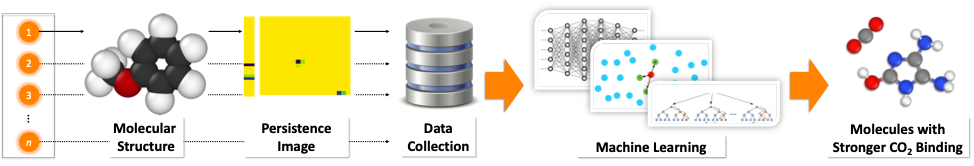
Related Publications:
(1) O. Queen, G. A. McCarver, S. Thatigotla, B. P. Abolins, C. L. Brown, V. Maroulas, K. D. Vogiatzis Polymer Graph Neural Networks for Multitask Property Learning, npj Comput. Mater., 2023, 9, 90.
(2) J. Townsend, P. Micucci, J. H. Hymel, V. Maroulas, K. D. Vogiatzis, Representation of Molecular Structures with Persistent Homology for Machine Learning Applications in Chemistry, Nat. Commun., 2020, 11, 3230.
(3) J. Townsend, N. M. Braunscheidel, K. D. Vogiatzis, Understanding the Nature of Weak Interactions Between Functionalized Boranes and N2/O2, Promising Functional Groups for Gas Separations, J. Phys. Chem. A, 2019, 123, 3315.
(4) C. R. Maroon, J. Townsend, K. R. Gmernicki, D. J. Harrigan , B. J. Sundell, J. A. Lawrence, III, S. M. Mahurin, K. D. Vogiatzis, B. K. Long, Elimination of CO2/N2 Langmuir Sorption and Promotion of “N2-Phobicity” within High-TgGlassy Membranes, Macromolecules, 2019, 52, 1589.
3. Computational Catalysis
Fixation of small molecules such as CH4, N2, and CO2 involves the cleavage of strong bonds, reservoirs of chemical energy, and its successful utilization depends on surmounting often large kinetic barriers. Nature has developed efficient enzymes that convert these molecules under mild conditions. It has long been recognized that metal ions can reduce these barriers by binding and activation processes. Cooperative effects between metal-metal, and/or metal-ligand can enhance the performance of the catalytic centers and achieve the conversion of CH4, N2, and CO2 in milder conditions and with cheap, earth-abundant metals.
We are interested on the theoretical examination of biomimetic active sites that are either supported on nanomaterials (eg. metal-organic frameworks, zeolites) or incorporated in model molecular complexes. Our studies are currently focused on metal-oxo sites, such as heme and non-heme Fe(IV)-oxo groups and CuxOy sites.
Related Publications:
(1) G. M. Jones, B. A. Smith, J. K. Kirkland, K. D. Vogiatzis Data-Driven Ligand Field Exploration of Fe(IV)-oxo Sites for C-H Activation, Inorg. Chem. Front., 2023, 10, 1062.
(2) G. A. McCarver, T. Rajeshkumar, K. D. Vogiatzis, Computational catalysis for metal-organic frameworks: An overview, Coord. Chem. Rev., 2021, 436, 213777.
(3) D. M. Shakya, O. A. Ejegbavwo, T. Rajeshkumar, S. D. Senanayake, A. J. Brandt, S. Farzandh, N. Acharya, A. M. Ebrahim, A. I. Frenkel, N. Rui, G. L. Tate, J. R. Monnier, K. D. Vogiatzis, N. B. Shustova, D. A. Chen, Selective Catalytic Chemistry at Rhodium(II) Nodes in Bimetallic Metal–Organic Frameworks, Angew. Chemie Int. Ed., 2019, 58, 16533.
(4) K. D. Vogiatzis, M. V. Polynski, J. K. Kirkland, J. Townsend, A. Hashemi, C. Liu, E. A. Pidko, Computational Approach to Molecular Catalysis by 3d Transition metals: Challenges and Opportunities, Chem. Rev., 2019, 119, 2453.